1. Introduction
ANACONDA is a tool designed for detecting and annotating somatic copy number alterations (CNVs) by integrating four state-of-the-art CNV-calling methods: ExomeCNV, FREEC, ADTEx and EXCAVATOR. In the framework of ANACONDA, configurations of all methods and CNV-calling procedures are automatically done, which is very convenient for non-professional users. Furthermore, the common CNVs detected by multiple algorithms are extracted and further annotated in several dimensions using currently available resources including xxx. Finally, an html file is generated to provide a detailed display of the information about parameter settings, detected CNVs, annotation results and etc.
2. Requirements
√ Unix-like systems
√ R3.0+: The main analysis of CNVs in ExomeCNV, ADTEx and EXCAVATOR is done using R statistical language.(Note: R3.3 doesn’t support some needed packages, we have run ANACONDA successfully by R3.2.2 and R3.2.4. So we highly recommend R3.2.)
√ Jdk8+: Java language is required by ExomeCNV to derive depth of coverage
√ Other necessary tools: gcc and g++ to compile source code
3. Installation
The installation step prepares the running environments for the adopted CNV-calling methods including: 1) check if programing environments are ready, and 2) install dependent packages required by each method. We provide a script named “install” in the top directory of ANACONDA package to complete these steps, users can type following commands to install the software:
chmod +x install
./install
We suggest that users run above two commands as root user, otherwise it may throw an error of “permission denied”. During installation, any errors occurred are recorded in file “log.err”, users can check this file to find more details. After the installation, the main program of ANACONDA is generated in “bin” directory.
Note: Because R package ‘Hmisc’ has so many dependencies, users should install it manually. You can type following command in R shell to install it:
install.packages('Hmisc',dependencies=T,repos="http://cran.rstudio.com/")
4. Configuration
The configuration step setups all required parameters and provides inputs for each algorithm. Generally, the parameters are divided into five groups:
♦ General. The common parameters requited by multiple methods are specified in this part. These parameters including selected methods for calling CNVs, tumor/normal BAM file, the sequence file of reference genome, target capture file, tumor ploidy and etc.
♦ ExomeCNV. The specific parameters for ExomeCNV: (i) parameters for calling exon-level CNVs, and (ii) parameters for combing exons.
♦ FREEC. Parameter settings for FREEC. These parameters will be used to automatically generate the specific configuration file for FREEC
♦ ADTEx. The only one parameter needs to be specified for ADTEx, namely minimum read depth in control sample.
♦ EXCAVATOR. Parameters uniquely required by EXCAVATOR are listed in this part
We provide a detailed description of all parameters as follows:
| Group | Parameter | Description | Possible values |
|---|---|---|---|
| General | methods | Methods for CNV calling | Default: ExomeCNV, FREEC, ADTEx, EXCAVATOR Ex: ExomeCNV, ADTEx |
| tumor | Tumor BAM file | Ex: /path/to/tumor.bam | |
| normal | Normal BAM file | Ex: /path/to/normal.bam | |
| ref | Genome reference sequence file | Ex: /path/to/ref.fa | |
| refVersion | Version of human genome | Ex: hg18 or hg19 | |
| target | File with capture regions | Ex: /path/to/target.bed | |
| out | Output directory to save results | Ex: /path/to/outputDir | |
| ploidy | Tumor ploidy | Default: 2 Ex: 2 or 3 or 4 | |
| purity | Tumor purity | Default: 1.0 Ex: value between 0 and 1 | |
| cnaCalledMethods | A CNV is considered to be a common CNV if it is called by multiple methods | Default: 1 Ex: a value belongs to [1,2,…,#methods] | |
| geneMinCoverage | Minimum percentage of a gene to be covered by a CNV | Default: 0.7 Ex: value between 0.5 and 1.0 | |
| ExomeCNV | min.spec1 | Minimum specificity for calling exon-level CNVs | Default: 0.9999 Ex: value between 0 and 1 |
| min.sens1 | Minimum sensitivity for calling exon-level CNVs | Default: 0.9999 Ex: value between 0 and 1 | |
| option1 | Optimization Strategy for calling exon-level CNVs | Default: "spec" Ex: "spec" or "auc" | |
| min.spec2 | Minimum sensitivity for combining exons | Default: 0.99 Ex: value between 0 and 1 | |
| min.sens2 | Minimum sensitivity for combining exons | Default: 0.99 Ex: value between 0 and 1 | |
| Option2 | Optimization Strategy for combining exons | Default: "auc" Ex: "spec" or "auc" | |
| FREEC | BedGraphOutput | Set TRUE if you want an additional output in BedGraph format for the UCSC genome browser | Default: FALSE |
| breakPointThreshold | Positive value of threshold for segmentation of normalized profiles | Default: 0.8 | |
| breakPointType | Desired behavior in the ambiguous regions | Default: 2 | |
| chrFiles | Path to the directory with chromosomes fasta files | Ex: /path/to/hg18 | |
| contaminationAdjustment | Set TRUE to correct for contamination by normal cells | Default: FALSE | |
| forceGCcontentNormalization | Simply model "sample RC ~ Control RC" | Fixed value: 0 | |
| intercept | Intercept of polynomial | Fixed value: 0 | |
| minCNVlength | Minimal number of consecutive windows to call a CNV | Default: 3 | |
| maxThreads | Number of threads | Default: 1 | |
| degree | Degree of polynomial | Fixed value: 1 | |
| readCountThreshold | Threshold on the minimal number of reads per window in the control sample | Default: 10 | |
| sex | Sample sex | "sex=XX" will exclude chr Y from the analysis "sex=XY" will not annotate one copy of chr X and Y as a loss | |
| mateOrientation | Format of reads | Fixed value: 0 | |
| ADTEx | minReadDepth | Threshold on the minimum read depth in the control sample | Default: 10 |
| EXCAVATOR | wigFile | Mappability file | Ex: /path/to/hg18_uniqueome.Wig |
| mapQ | Mapping quality for BAM file filtering | Default: 0 | |
| Omega | Omega parameter for the HSLM algorithm | Default: 0.1 | |
| Theta | Theta parameter for the HSLM algorithm | Default: 1e-4 | |
| D_norm | D_norm parameter for the HSLM algorithm | Default: 10e6 | |
| Thre_d | Threshold d for the truncated gaussian distribution of the FastCall Calling algorithm | Default: 0.5 | |
| Thre_u | Threshold u for the truncated gaussian distribution of the FastCall Calling algorithm | Default: 0.35 | |
| minSeglen | Segment with a number of exons smaller than a threshold are filtered out | Default: 4 |
5. Running ANACONDA
Running the main program of ANACONDA only needs one argument that is the configuration file. Type following command to run ANACONDA:
./bin/ANACONDA /path/to/configfile
For example:
./bin/ANACONDA ./config/config.txt
6. Results summary
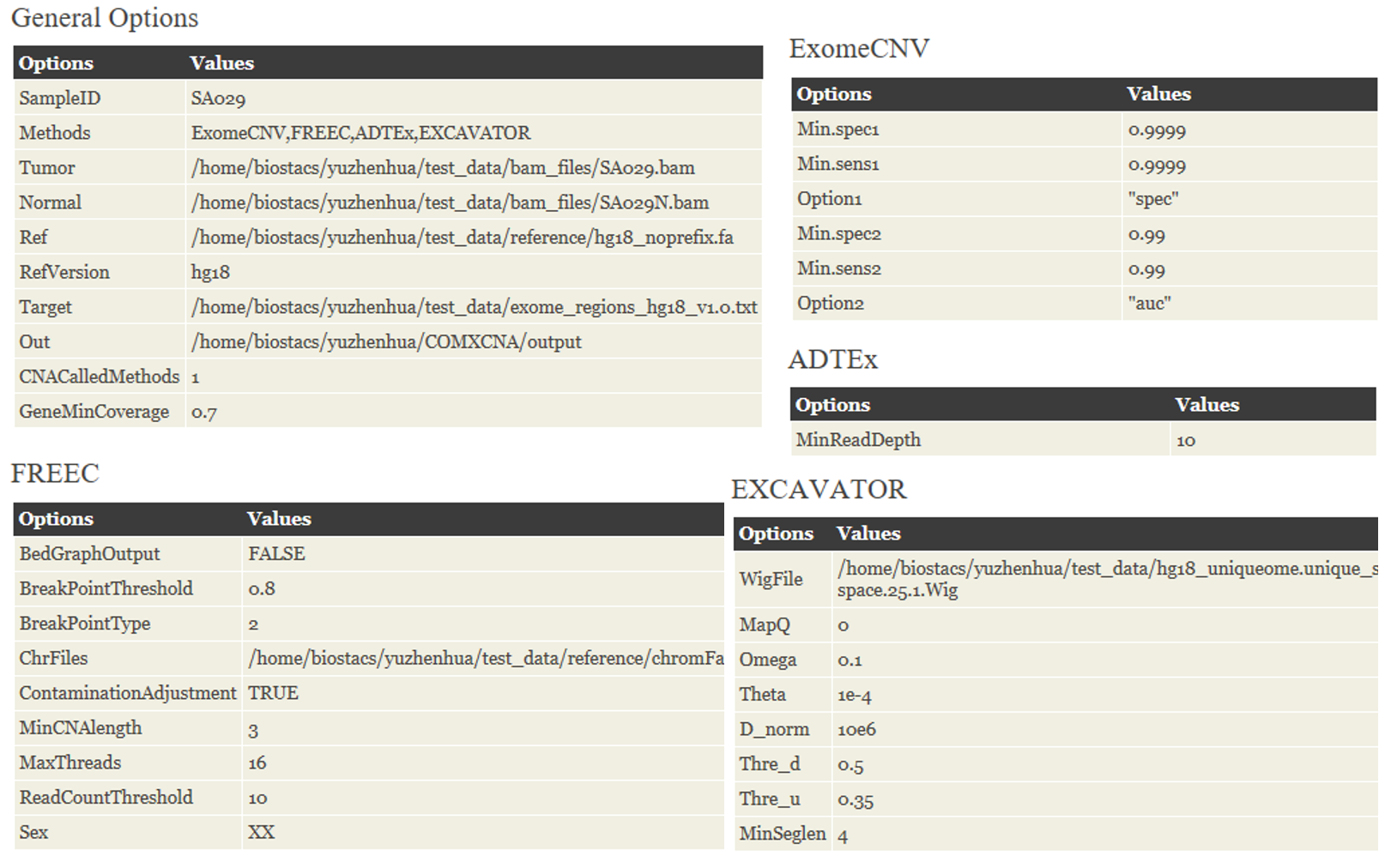
♦ Parameters for variant caller
♦ Detailed results for CNV caller

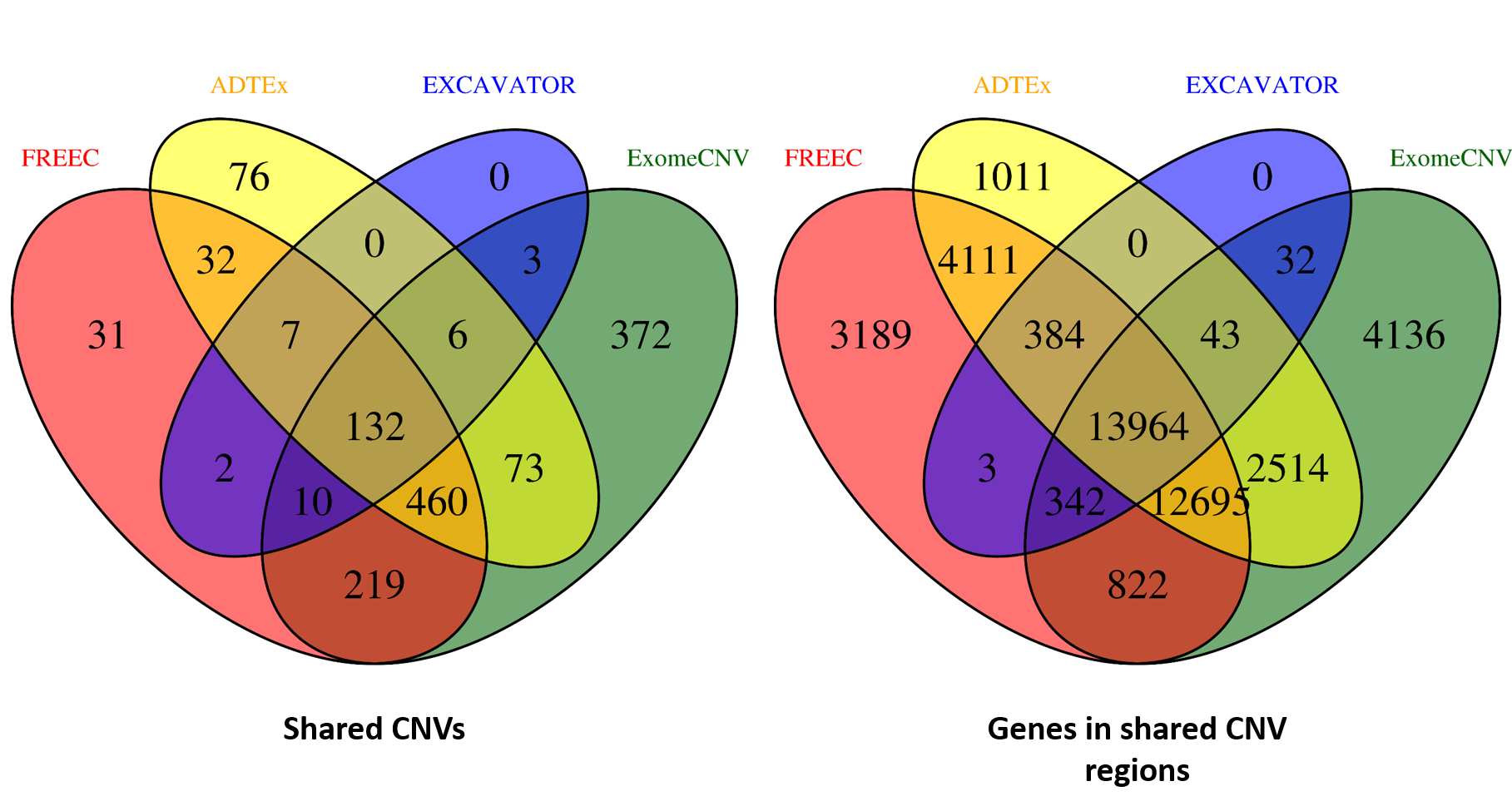
♦ Shared CNVs
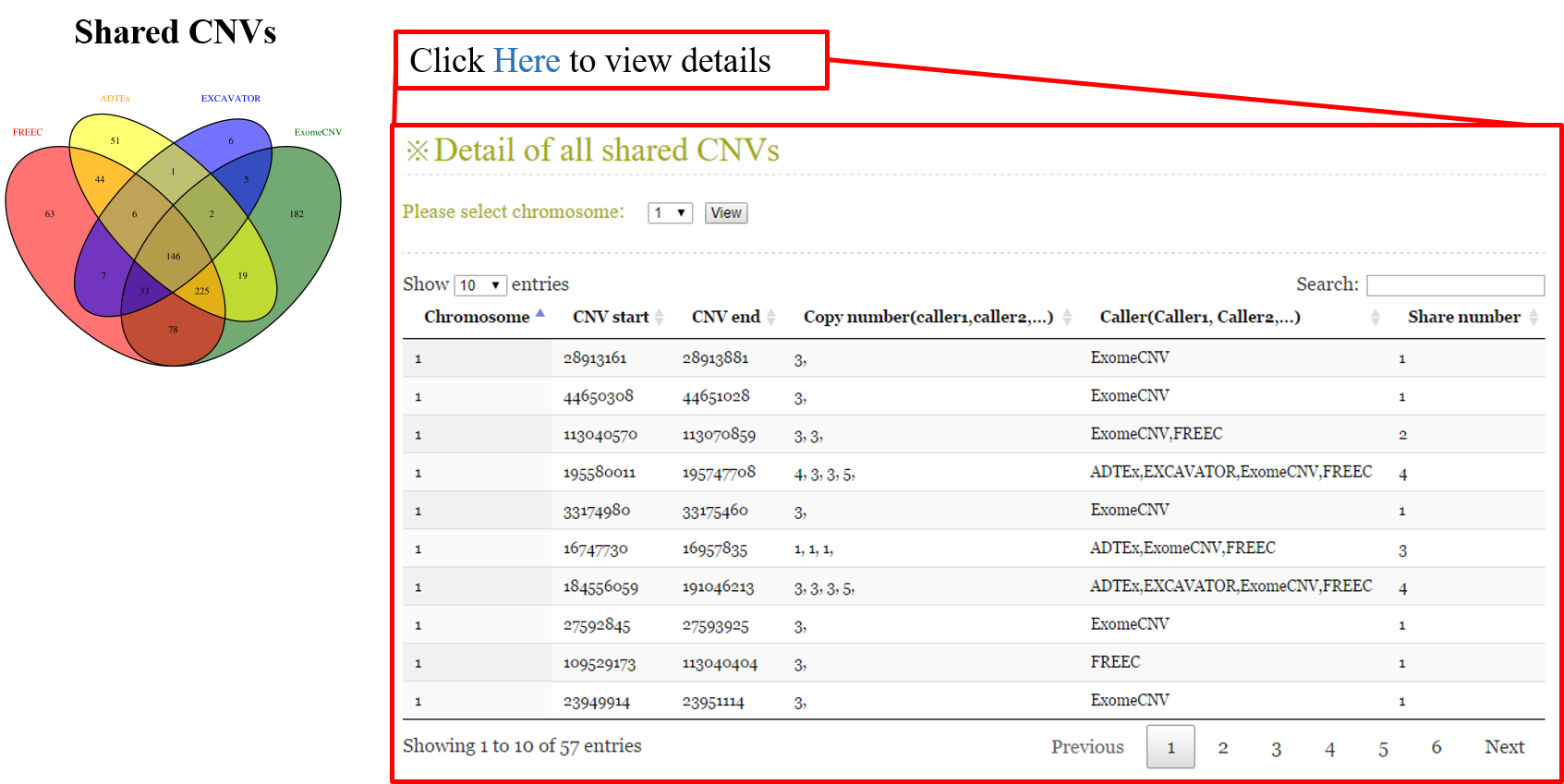
♦ Detail of shared CNVs
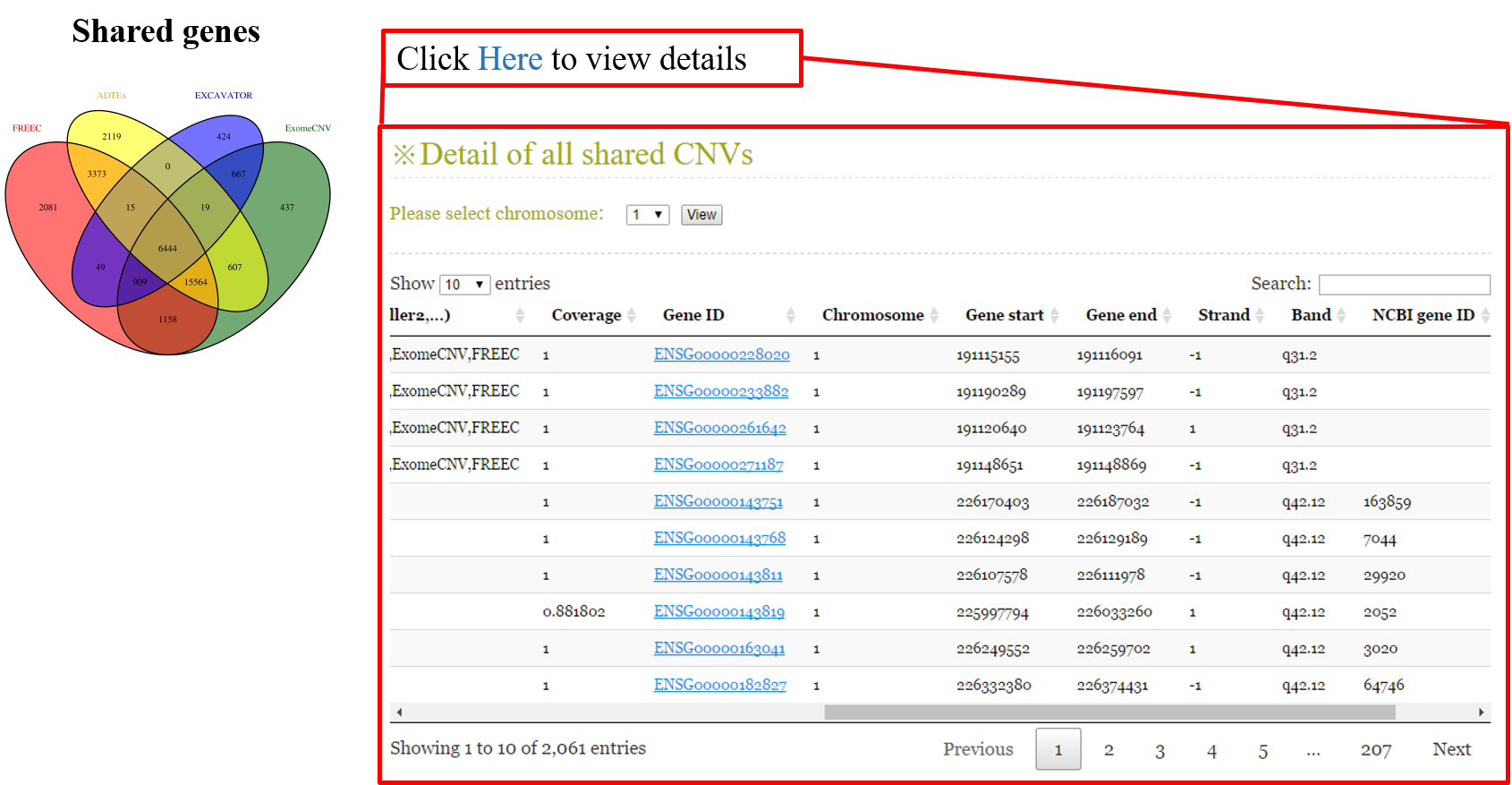
♦ Annotation
