CPSS HELP
For all users, CPSS can analyze the small RNA deep sequencing datain different
architecture: single and paired.
By using CPSS, small RNA NGS data can be analyzed systematically in one platform after a single submission of data by integration of annotation and functional analysis of novel and/or differentially expressed miRNAs. CPSS generates an analysis report including: 1) annotation analysis, which provides a comprehensive analysis for small RNA transcriptome, such as the length distribution and genome mapping of the sequencing reads, small RNA annotation, prediction of novel miRNAs, identification of differentially expressed miRNAs, piRNAs and other small RNAs between samples, and detection of miRNA SNPs and isoforms; 2) functional analysis, which provides the functional analysis of miRNAs, e.g. predicting miRNA target genes by multi-tools, enriching Gene Ontology terms (GO), performing signalling pathways, and protein-protein interaction (PPI) analysis for the predicted genes.
1. If users provide an email address, our server will send a reminder email to users when the job is done. Users could get the results after receiving the reminder email.

2. When uploaded to CPSS, the data should be in a specified file format (*.fa or *.gz). And a Seq-clean program was provided in CPSS package (http://mcg.ustc.edu.cn/bsc/sdap/sdap_tools.html), which could perform the transformation of raw data form Illumina Genome Analyzer, 454 FLX instrument. The FASTA format (*.fa) files can be further compressed into *.gz format using a software that can be found in http://www.7-zip.org/download.html for free. Several small RNA NGS data from different species are provided in CPSS, and users could download these data to test our web server. In addition, the maximum allowable size of data input is not limited..

3. Users should choose the reference genome through a scroll bar for sequence reads alignment. Currently, CPSS is used for the analysis of small RNA deep sequencing data from ten organisms, and will be extended to the analysis of data from more species in near future.

4. All the reads were classified into two groups, mapped and unmapped tags, based on whether they can be mapped onto the reference genome using the SOAP2 program. The mapped tags are then aligned against miRBase, Rfam, repeat database, and the coding region of reference genome, and be grouped as miRNA, other small RNA, mRNA, genomic repeats, or unclassified reads if they can not be assigned to any of the above groups. For detection of piRNAs from NGS data, all know piRNA sequences were download from NCBI Nucleotide database for three species (Homo, Mus, and Rattus), and the index was created by makeblastdb in Blast 2.2.5 package. And the genome mapped sequences were matched to the piRNA index by blastn.
Parameters for SOAP2.
M: Match mode 0:exact match 2:1 mismatch 3:all 4:best hits.
v: Maximum number of mismatches.
r: Report repeat hits 0:none 1:random one.
Parameters for BlastN.
E: Blast e-value.

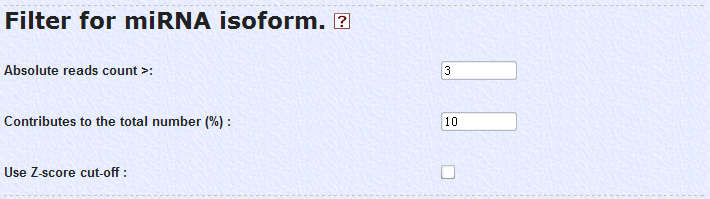
5. To identify miRNA isoforms for the known miRNAs from NGS data, we applied several filters in the analysis packages. First, the reads classified into isoforms must be numbered above 3 times in the data. Second, every isoform considered in the analysis must contribute in more than 10% to the total number of all tags annotationed into the same miRNA. Third, we used Z-score option to exclude the sequencing errors
6. Users could choose mireap or miRdeep to predict novel miRNAs for small RNA NGS data from differrnt species.

7. In the default settings of CPSS, the ten most abundant novel miRNAs and known miRNAs will be subjected to target prediction for a single sample. In these miRNA prediction tools, miRanda and RNAhybird could predict the targets for known and novle miRNAs based on miRNA sequences, however, other six tools only predict the targets for known miRNAs. In CPSS, users must select one of miRanda and RNAhybird at least for miRNA target prediction. And users could perform the united application of these tools to improve accuracy of prediction, or intersection of these tools to cover positive targets.
Chromosome distribution of all sequences and unique sequences of the sample.
Parameters for miRanda to predict the targets.
Energy: The minimal free energy for miRanda.
Score: The minimal score for miRanda.
Parameters for RNAhybird to predict the targets.
Energy: The minimal free energy for RNAhybird.
P value: The basis for judgment of RNAhybird.

8. For functional annotation of miRNAs, the miRNA predicted target genes are subjected to the analysis of Gene Ontology terms. In the default settings of CPSS, a GO enriched gene will be matched to the signalling pathway annotation dataset downloaded from KEGG.
Parameters for GO analysis of miRNA's target.
P value: The threshold for hypergeometric test.
Enrichment fold: Enrichment fold of GO terms.
Parameters for Pathway analysis.
P value: The threshold for hypergeometric test.
Enrichment fold: Enrichment fold of Pathway name.

9. Genes enriched in key GO terms are used for PPI analysis. CPSS will map the enriched genes to the String database.
Parameters for PPI analysis.
Score: The total score for a given PPI record provided by String database.
